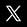|
|
|
|
 |
|
oxydoréduction |
|
|
| |
|
| |
oxydoréduction
Consulter aussi dans le dictionnaire : oxydoréduction
Cet article fait partie du dossier consacré à la réaction chimique.
image: http://www.larousse.fr/encyclopedie/data/animations/1100596-Oxydor%c3%a9duction_c%c5%93ur_de_mercure.jpg
Oxydoréduction, cœur de mercure
image: http://www.larousse.fr/encyclopedie/data/vignettes/1100599.jpg
Pièce de cuivre qui se couvre d'argent puis d'or
Action chimique d'un corps oxydant sur un corps réducteur, avec à la fois une oxydation du réducteur et une réduction de l'oxydant.
Le phénomène d’oxydoréduction
L'importance de l'oxydoréduction, phénomène chimique ou électrochimique, est considérable tant par ses effets spontanés, bénéfiques (photosynthèse) ou néfastes (corrosion), que par ses applications multiples dans la vie quotidienne (piles, accumulateurs…) ou dans l'industrie chimique et électrochimique.
Depuis la découverte du feu, l'homme a su mettre à profit la combustion du bois pour obtenir de l'énergie thermique ainsi que la réduction des minerais lors de la préparation du fer, sans connaître les réactions chimiques mises en jeu. Ce n'est qu'à la fin du xviiie s. que la combustion est apparue comme une oxydation, c'est-à-dire une combinaison avec un élément de l'air, l'oxygène.
Au sens strict, l'oxydation est la fixation d'oxygène sur un corps, la réduction, le phénomène inverse ; ces deux phénomènes sont d'ailleurs généralement couplés. Ainsi, dans la réaction 2CuO + C → 2Cu + CO2, on assiste simultanément à l'oxydation du carbone et à la réduction de l'oxyde de cuivre ; ce dernier est l'oxydant (noté Ox), le carbone étant le réducteur (Red) ; la réaction est une oxydoréduction.
Plusieurs éléments, tels les halogènes, le soufre, etc., se comportent dans certaines de leurs réactions comme l'oxygène ; on convient de dire que, par exemple, la combustion du sodium dans le chlore (qui donne Na+Cl−) est une oxydation du métal, au même titre que sa combustion dans l'oxygène. La transformation du sodium en ions Na+ est une oxydation du sodium ; elle est obtenue par enlèvement d'électrons. Corrélativement, la réduction du chlore en ions chlorure Cl− résulte de la fixation d'électrons. Au cœur de l'oxydoréduction se trouve donc l'électron, particule très mobile et omniprésente dans toutes les formes de la matière.
Le couple redox
Le sens du terme oxydoréduction, contraction de « oxydation » et de « réduction », a évolué au cours du temps : ainsi pour Antoine Laurent de Lavoisier, le terme oxydation signifiait fixation d'oxygène, et le mot réduction désignait la perte d'oxygène, par exemple :
S + O2 → SO2 (oxydation),
2HgO → 2Hg + O2 (réduction).
Cependant, on constate que dans de nombreux cas l'expression ne porte que sur l'évolution chimique d'un seul des deux constituants de la réaction ; ainsi en est-il de la réduction de l'oxyde de cuivre II par le dihydrogène :
CuO + H2 → Cu + H2O,
où une ambiguïté apparaît, dans les termes, car on néglige ici le dihydrogène qui, lui, subit une oxydation. En fait, et d'une manière générale, toute réaction de réduction est accompagnée d'une réaction d'oxydation, et réciproquement. Seule l'électrochimie permet de les séparer dans certaines conditions.
Mais il apparaît que la notion d'oxydation peut s'appliquer aussi à la réaction :
Li + 1/2 Cl2 → LiCl,
comparable à la réaction :
2Li + 1/2 O2 → Li2O ;
dans la première, le lithium (Li) est transformé en Li+ par la perte d'un électron (e−), récupéré par le dichlore ; dans la seconde, deux électrons perdus par deux atomes Li sont fixés par l'oxygène pour former l'ion O2− ; dans les deux cas, on dit qu'il s'agit d'une oxydation du lithium due à une perte d'électrons, accompagnée d'une réduction du chlore ou de l'oxygène due, elle, à un gain d'électrons.
Une autre façon d'exprimer le phénomène est de dire que le lithium est un réducteur, c'est-à-dire une espèce chimique pouvant fournir un électron, et que le dichlore et le dioxygène sont des oxydants susceptibles de capter un ou des électrons. Li et Li+ forment ce qu'on appelle un couple redox (abréviation de réduction-oxydation), noté Li+/Li (soit Ox/Red).
On l'écrit : Li ⇌ Li+ + e−,
Li étant le réducteur, et Li+ l'oxydant. La double flèche indique que, dans certaines conditions, on sait réaliser :
Li+ + e− → Li (ce qui est du domaine de l'électrochimie).
D'une manière générale, on peut écrire les demi-réactions d’oxydoréduction suivantes :
Red1 ⇌ Ox1 + n1e−
et :
Ox2 + n2e− ⇌ Red2.
Les réactions d'oxydoréduction
Les équations chimiques correspondant aux réactions d'oxydoréduction peuvent être décrites en utilisant les couples redox, sous réserve de satisfaire à certaines conditions thermodynamiques, et de telle sorte que les électrons fournis par le réducteur 1 (Red1) soient récupérés par l'oxydant 2 (Ox2). Ainsi, l'équation chimique générale d'oxydoréduction :
n2Red1 + n1Ox2 ⇌ n2Ox1 + n1Red2
fait-elle intervenir les deux couples redox suivants :
(Red1 ⇌ Ox1 + n1e−) . n2
et
(Ox2 + n2e− ⇌ Red2) . n1,
dont l'addition permet de retrouver l'équation précédente. Cette façon de procéder est aisée quand interviennent des réactions ioniques qui permettent de connaître n1 et n2 sans difficulté. Par exemple, l'action d'un acide en milieu aqueux sur le zinc fait intervenir deux couples redox bien connus :
Zn → Zn2+ + 2e−,
(H3O+ + e− → 1/2 H2 + H2O) . 2.
Dans les conditions thermodynamiques convenables, les deux réactions se produisent dans le sens 1, donnant la réaction globale :
Zn + 2 H3O+ → Zn2+ + H2 + 2 H2O.
Le degré d'oxydation
Il existe aussi des couples redox tels que :
Fe2+ ⇌ Fe3+ + e−, où l'on voit aisément que Fe3+ est plus oxydé que Fe2+, puisqu'il résulte de la perte d'un électron par ce dernier.
Mais, avec des ions comme Cl−, ClO−,ClO3−, ClO4−, il est difficile de distinguer les éléments les plus oxydés.
Les chimistes ont été ainsi amenés à attribuer un degré d'oxydation, ou nombre d'oxydation (NO), positif ou négatif, aux différents éléments et à leurs ions lorsqu'ils sont engagés dans divers composés ; pour le distinguer de la charge des ions, on le représente par un chiffre romain, précédé du signe + ou −. Pour déterminer ce nombre formel, on utilise les règles suivantes :
– tout élément à l'état de corps pur a un nombre d'oxydation 0 (c'est le cas de O dans O2, de H dans H2, de Fe dans le fer métal) ;
– dans une espèce chimique non chargée, la somme algébrique des NO des éléments constitutifs est égale à 0 (c'est le cas pour H et O dans H2O, pour Na et Cl dans NaCl);
– dans une espèce ionique, la somme algébrique des NO des éléments constitutifs est égale à la charge de l'ion. Ainsi, dans ClO−, la somme algébrique des NO de Cl et de O est −1 ; il en est de même dans ClO4−.
Partant de ces règles, le NO d'un élément dans une molécule ou dans un ion est, en valeur absolue, égal au nombre d'électrons qu'il aurait fixés, si c'est l'élément le plus électronégatif, ou qu'il aurait perdus, si c'est l'élément le moins électronégatif, si on considérait que toutes ses liaisons sont ioniques. On lui attribue le signe − s'il a fixé les électrons, le signe + s'il les a perdus. Plus le nombre d'oxydation est élevé (en valeur algébrique), plus l'élément est oxydé ; plus il est bas, plus l'élément est réduit. Par exemple :
H+, Li+, Na+, … NO = + I ;
Al3+, Te3+, Cr3+, … NO = + III ;
H−, F−, Cl−, … NO = − I ;
O2−, S2−, … NO = − II.
Dans le cas des ions simples, le NO est donc égal à la charge de l'ion.
L'équation de Nernst
Si le système redox :
Ox + ne− ⇌ Red,
obéit aux lois de la thermodynamique (les phénomènes cinétiques, très importants dans ce domaine, risquent de masquer le comportement thermodynamique), une expression relativement simple, l'équation de Nernst, lie le potentiel ET de ce système (où la concentration de l'oxydant et celle du réducteur ne sont pas égales à 1 mol . −1) au potentiel standard E0T (où la concentration de l'oxydant et celle du réducteur sont égales à 1 mol . l−1) et au nombre n d'électrons échangés à une température donnée T :
ET = E0T + (RT/nΦ) × log [Ox]/[Red],
où E0T, potentiel standard du système (sous une pression de 1 bar), est une constante, consignée dans des tables, qui en caractérise la nature chimique à une température donnée T exprimée en degrés Kelvin (si T = 298 K, E0 est le potentiel standard normal),
R la constante des gaz parfaits (8,31 J . K−1 . mol−1),
Φ la constante de Faraday (96 500 C . mol−1),
[Ox] et [Red] correspondant aux activités de Ox et de Red que l'on assimile ici aux concentrations [Ox] et [Red] en mol . l−1.
À 298 K (25 °C), cette expression s'écrit souvent sous la forme :
ET = E0T + (0,06/n) × log [Ox]/[Red],
où les potentiels sont exprimés en volts (V).
Prévision des réactions
En solution, lorsque l’on a en présence deux couples rédox Ox1/Red1 et Ox2/Red2, de potentiels standards respectifs E01 et E02, et que E01 > E02, c'est l'oxydant 1, appartenant au couple redox 1 – dont le potentiel est le plus élevé – qui oxyde le réducteur 2 appartenant au couple redox 2 – dont le potentiel est le moins élevé.
Applications
Les réactions d'oxydoréduction interviennent dans nombre de processus naturels ou artificiels, tant dans des réactions en solution qu'en phase solide. Tel est le cas de la plupart des réactions métallurgiques permettant l'élaboration des métaux à partir de leurs minerais, comme celle citée en introduction. Ces réactions, dont certaines étaient connues depuis l'Antiquité, mettaient déjà en pratique des principes thermodynamiques qui ne furent élucidés que bien plus tard. Cela explique que, au cours de l'histoire, les métaux ont été élaborés dans l'ordre de difficulté croissante de réduction, depuis le cuivre, vers 4000 avant J.-C., jusqu'à l'aluminium, en 1825. Les phénomènes d'oxydoréduction ont également un rôle crucial en biologie. Ils permettent notamment la respiration cellulaire des organismes vivants.
DOCUMENT cnrs LIEN
|
| |
|
| |
|
|
|
Les promesses du contrôle quantique |
|
|
| |
|
| |
LES PROMESSES DU COTRÔLE QUANTIQUE
Contrôler les réactions chimiques par laser, manipuler les bits d’un ordinateur quantique, améliorer les images des IRM… Voilà ce que promet le contrôle quantique, une discipline en plein boom qui s’apprête à révolutionner notre quotidien.
Laser, microprocesseur, GPS, imagerie par résonance magnétique… Aujourd’hui, sans qu’on s’en rende compte, de nombreuses techniques utiles dans la vie de tous les jours n’auraient pas pu voir le jour sans la mécanique quantique, une théorie inventée dans les années 1920 qui décrit les phénomènes physiques à l’échelle des atomes. « Mais aussi importantes soient-elles, ces applications s’appuient seulement sur une compréhension passive des lois quantiques, note Dominique Sugny, du Laboratoire interdisciplinaire Carnot de Bourgogne1, à Dijon, et de l’Institute for Advanced Study de Munich, en Allemagne. Désormais, les chercheurs veulent aller plus loin, en contrôlant activement les objets à cette échelle. » Discipline en plein développement, le contrôle quantique, qui vise à manipuler des atomes, des photons ou autres électrons afin de leur faire réaliser des tâches bien déterminées, pourrait, dans les années qui viennent, révolutionner plus encore notre quotidien.
Une méthode inspirée par l’aéronautique
Le contrôle quantique a fait ses balbutiements dans les années 1980. Les physiciens et les chimistes rêvaient alors de pouvoir contrôler les réactions chimiques par laser. L’idée étant que, lorsque l’on éclaire une molécule avec un laser, celui-ci y dépose de l’énergie, ce qui met en mouvement les noyaux atomiques et les électrons, impliqués dans les liaisons chimiques. Si la fréquence du rayonnement correspond exactement à celle à laquelle vibrent les liaisons chimiques, on peut alors « exciter » des liaisons qui se retrouvent fragilisées et se cassent plus facilement. Certaines réactions, plutôt que d’autres, se voient ainsi favorisées.
Les premières expériences ne tardent pas à se mettre en place. Mais rapidement, on s’aperçoit que l’exercice est beaucoup plus compliqué que prévu. Car, en pratique, l’énergie déposée sur une liaison se répartit vite sur l’ensemble de la molécule. Au point que l’on ne maîtrise plus grand-chose. « Les premiers essais de contrôle par laser relevaient plutôt de méthodes empiriques reposant sur des arguments qualitatifs, confie Dominique Sugny. Restait encore à mettre au point une stratégie de contrôle efficace. »
Pour y parvenir, les physiciens et les chimistes s’inspirent alors d’une théorie développée trente ans plus tôt pour décrire les objets macroscopiques du monde classique : la théorie du contrôle optimal. Celle-ci consiste à rechercher une solution sous la forme mathématique d’un problème d’optimisation qu’on peut résoudre numériquement. C’est ce principe qui a été utilisé, par exemple, dans les années 1960 en aéronautique militaire pour optimiser les trajectoires de montée en altitude des avions de chasse, où il a permis de diviser par deux les temps de montée avec des trajectoires contraires à l’intuition. Ou encore au cours du programme lunaire américain Apollo pour trouver les trajectoires minimisant le plus possible la consommation de carburant pour atteindre notre satellite naturel.
Trajectoire de la mission Apollo 11 Illustration montrant la trajectoire et les différentes étapes de la mission Apollon 11, durant laquelle Neil Armstrong et Buzz Aldrin ont marché sur la Lune le 20 juillet 1969.
C. LUNAU/SCIENCE PHOTO LIBRARY/COSMOS
Partager
Maîtriser l’étrangeté quantique
Le contrôle quantique n’est donc pas qu’une histoire de physiciens et de chimistes. Ces derniers doivent s’entourer également de mathématiciens pour concevoir des algorithmes de calcul performants. Mais transposer les idées du contrôle optimal du monde classique au monde quantique s’est révélé une tâche ardue. « Il aura fallu attendre les années 2000, et le développement de moyens de calculs suffisamment puissants, pour atteindre cet objectif », souligne Dominique Sugny, membre du projet européen Quaint, qui met en réseau 17 laboratoires du Vieux Continent travaillant sur le contrôle quantique.
Les équations
décrivant
les phénomènes
quantiques
sont autrement
plus complexes
que celles régissant
les systèmes
classiques.
Il faut dire que les équations décrivant les phénomènes quantiques sont autrement plus complexes que celles régissant les systèmes classiques. Cela vient du fait que les particules microscopiques possèdent des propriétés incompatibles avec celles des objets macroscopiques. Première propriété déroutante : la superposition d’états. Prenons un électron. Celui-ci ne se trouve pas vraiment ici ou là autour du noyau de l’atome mais à plusieurs endroits à la fois. Et il ne se déplace pas à une vitesse donnée mais à plusieurs vitesses à la fois. Ce n’est qu’en effectuant une mesure sur l’électron que celui-ci apparaît alors à une position et à une vitesse bien précise. Avant toute mesure, les physiciens doivent donc considérer que l’électron est simultanément dans plusieurs états possibles.
Autre propriété déconcertante : l’intrication. Deux objets quantiques de même nature, des photons par exemple, peuvent former un seul et même système. Leurs états physiques sont alors corrélés. Quelle que soit la distance qui sépare ces deux objets, une mesure sur l’un affecte l’autre de façon instantanée. Ce sont précisément ces deux effets – la superposition et l’intrication – que le contrôle quantique tente d’exploiter de la manière la plus efficace possible.
Source de photons intriqués utilisée lors d'expériences de physique quantique Source de photons intriqués utilisée lors d'expériences de physique quantique.
M. BREGA/LOOK AT SCIENCES
Partager
Contrôler les réactions chimiques
Disposant d’une méthode générale pour contrôler leurs systèmes quantiques, les scientifiques font alors des pas de géant, d’abord dans le contrôle des réactions chimiques par laser. Les simulations numériques permettent ainsi d’identifier l’onde adéquate à envoyer sur la molécule : cette impulsion lumineuse doit comporter des modulations significatives d’amplitude et de fréquence sur des temps extrêmement courts – de quelques dizaines de femtoseconde, 10–15 secondes – comparables aux périodes de vibrations propres de la molécule.
En juin,
des chimistes
ont réussi à créer
une liaison entre
deux atomes
de magnésium.
Une première.
Aujourd’hui, grâce au contrôle par laser, les chimistes parviennent à casser préférentiellement des liaisons sur des molécules de plus en plus complexes. Et ils ont même réussi, en juin dernier, non pas à détruire, mais à créer une liaison entre deux atomes de magnésium2. Une première. Et une étape cruciale si l’on veut pouvoir un jour contrôler des réactions chimiques d’un bout à l’autre de la chaîne.
Manipuler les mémoires quantiques
Il est un autre domaine de recherche où l’on attend énormément du contrôle quantique : l’information quantique. Cette discipline vise à développer des moyens de calcul et de communication utilisant la logique quantique. Le Graal est la mise au point d’un ordinateur quantique, qui serait capable de résoudre des problèmes inaccessibles aux ordinateurs traditionnels.
Dans un ordinateur classique, l’information élémentaire, le bit, ne peut prendre que deux valeurs, 0 ou 1, selon le passage ou non du courant électrique à travers un transistor. Mais, dans un ordinateur quantique, le bit quantique (ou qubit), qui repose sur l’état d’un système quantique (atome, ion, photon, circuit supraconducteur…), peut, du fait de la superposition d’états, se retrouver dans ces deux états en même temps. De plus, lorsque deux qubits interagissent, leurs états physiques s’enchevêtrent, du fait de l’intrication.
Grâce à ces deux phénomènes, un ordinateur quantique peut, en théorie, avoir accès à la totalité des résultats en une seule étape quand un ordinateur classique doit traiter l’information un résultat après l’autre. Ce qui permettrait de réduire considérablement les temps de calcul.
Mais on est loin encore de la réalisation d’un tel ordinateur. Aujourd’hui, les physiciens ne parviennent à coupler au mieux qu’une vingtaine de qubits entre eux. Or, pour augmenter les capacités de calcul d’un ordinateur quantique, il faut manipuler bien plus de qubits. Le problème, c’est que cela augmente les chances de ces derniers d’interagir avec leur environnement (lumière parasite, champ magnétique, etc.) et de perdre alors leurs propriétés quantiques. Ce phénomène, baptisé décohérence, est le principal obstacle à la mise au point d’ordinateurs de ce type.
Le contrôle quantique vise justement à pallier cet effet néfaste. Comment ? En détectant les erreurs dues à la décohérence et en les corrigeant en temps réel. Un exercice qui semble impossible en théorie, puisque la mécanique quantique stipule que, pour mesurer une particule, il faut que celle-ci interagisse avec un détecteur matériel et donc se détruise – du fait de la décohérence.
Cavité Piège à photons Serge Haroche et Igor Dotsenko examinant une cavité "piège à photons".
C. LEBEDINSKY/CNRS PHOTOTHEQUE
Partager
Mais les chercheurs ne manquent pas d’imagination pour lever ces obstacles et, en 2011, une avancée majeure a été accomplie par l’équipe de Serge Haroche, du Laboratoire Kastler Brossel3, à Paris, Prix Nobel de physique en 2012. Le groupe de physiciens est parvenu à stabiliser le nombre de photons piégés dans une cavité supraconductrice4. Au préalable, les chercheurs avaient réussi à mesurer les photons sans les détruire. Et cette mesure avait généré un signal micro-onde qui réintroduisait un photon à l’instant approprié. Cette prouesse laisse entrevoir la possibilité de corriger un jour en temps réel les bits d’un ordinateur quantique. Même si les défis théoriques et expérimentaux demanderont encore beaucoup de travail pour être surmontés.
Améliorer la résolution des IRM
Autre champ d’application du contrôle quantique : l’imagerie par résonance magnétique (IRM). Si cet outil permet déjà d’aboutir à des diagnostics très précis, le contrôle quantique pourrait permettre d’améliorer plus encore ces derniers. L’IRM repose sur le principe de la résonance magnétique nucléaire : placés dans un champ magnétique, certains noyaux d’atomes peuvent absorber de l’énergie avant de la relâcher. L’énergie mise en jeu lors de ce phénomène de résonance correspond à une fréquence très précise qui dépend du champ magnétique appliqué et de l’environnement chimique de ces noyaux – les autres noyaux voisins et les électrons. En mesurant la fréquence de résonance, on peut remonter à la nature de la molécule et à sa position dans l’espace. Et ainsi obtenir une image des tissus et des organes.
Or ces images sont d’autant plus contrastées – et révèlent ainsi plus de détails – que le champ magnétique est soumis à des variations spécifiques d’amplitude et de fréquence. En utilisant la théorie du contrôle optimal, toujours elle, une équipe de chercheurs, dont Dominique Sugny, est parvenue à déterminer les profils de champ magnétique à appliquer pour obtenir le meilleur contraste possible d’une image pour des conditions expérimentales données. Les physiciens ont validé ensuite avec succès cette approche par une expérience test in vitro où ils ont pu distinguer dans une solution organique une zone reproduisant du sang sous sa forme oxygénée et une autre sous sa forme désoxygénée5
IRM à l'hôpital de la Pitié Salpêtrière IRM à l'hôpital de la Pitié Salpêtrière.
OPDT/BSIP
Partager
La prochaine étape sera d’appliquer ces concepts in vivo. « Nous espérons y parvenir d’ici à cinq ans », s’enthousiasme Dominique Sugny. Si ces tests se révélaient concluants, ils apporteraient un progrès considérable en matière d’IRM. Car ils permettraient de limiter l’utilisation des agents de contraste, une technique chimique d’amélioration de l’image pouvant présenter des risques pour le patient. On n’a pas fini de parler des bienfaits du contrôle quantique.
Notes
1. Unité CNRS/Univ. de Bourgogne.
2. « Coherent Control of Bond Making », L. Levin et al., Phys. Rev. Lett., 2015, vol. 114, 233003.
3. Unité CNRS/ENS/UPMC/Collège de France.
4. « Real-time Quantum Feedback Prepares and Stabilizes Photon Number States », C. Sayrin et al., Nature, 2011, vol. 477 : 73–77.
5. « Exploring the Physical Limits of Saturation Contrast in Magnetic Resonance Imaging », M. Lapert et al., Nature, Scientific Reports 2, 2012, vol. 589.
DOCUMENT cnrs le journal LIEN
Inscrivez le titre complet dans la rubrique " cnrs le journal "
|
| |
|
| |
|
|
|
LA CHIMIE DES SOLS |
|
|
| |
|
| |
LA CHIMIE DES SOLS
Le sol est un milieu essentiel pour de nombreux êtres vivants, il constitue un véritable réacteur biologique dont les principaux processus concernent la transformation de la matière organique. Du fait des pratiques culturales modernes, le taux de matière organique des sols tend à diminuer. La matière organique joue pourtant un rôle considérable sur les propriétés chimiques et physiques des sols par exemple la perméabilité, la stabilité structurale, la capacité de rétention et de circulation de l'eau. Le problème actuel grave de l'érosion des sols est pour une grande part une conséquence de teneurs insuffisantes en carbone organique. Mieux connaître les processus de fonctionnement des sols pour mieux les utiliser et les préserver impose de bien connaître leur matière organique et les réactions de transformation qui l'affectent. Dans les sols, les formes les plus intéressantes de matière organique sont les lipides et les substances humiques dont la structure moléculaire est encore largement inconnue et la définition peu précise. Le carbone organique des sols est d'origine végétale ou microbienne. L'activité biologique transforme une partie du carbone végétal ou microbien - par un processus naturel appelé humification - en composés de type " humique ", plus stables que les formes initiales et qui correspondent probablement à des formes transitoires de " stockage " de carbone organique dans les sols. Ces processus ne sont que partiellement connus. À terme, une partie du carbone organique-déchet " propre " pourrait être facilement recyclée, à un coût réduit économiquement acceptable, pour corriger les pratiques actuelles et ainsi compenser les déficits en carbone. Des résultats particulièrement intéressants ont déjà été obtenus. À l'inverse, certains sols carencés à activité biologique réduite, où la matière organique tend à s'accumuler, peuvent être facilement améliorés en compensant les carences et en favorisant ainsi le turn-over de la matière organique.
Texte de la 234ème conférence de l’Université de tous les savoirs donnée le 21 août 2000.La chimie des solspar André Amblès Le sol est un milieu essentiel pour de nombreux êtres vivants, il constitue un véritable réacteur biologique dont les principaux processus concernent la transformation de la matière organique. Il joue également, hélas, un rôle d'interface dans tous les phénomènes de pollution de notre environnement. La matière organique qu’il contient reste encore mal connue des hommes, les travaux significatifs concernant sa structure n’ayant débuté qu’il y a environ vingt cinq ans. Les recherches menées dans le domaine ont un intérêt fondamental certain, mais également un intérêt appliqué. En effet, l’accroissement de la population mondiale, le développement des activités humaines influencent de plus en plus le fonctionnement et les propriétés des sols. Pour des intérêts purement économiques, les pays industrialisés ont favorisé le développement de l’agriculture intensive, la réduction de la diversité des pratiques culturales et l’abus de la monoculture. Les engrais minéraux sont de plus en plus utilisés au détriments des fumures ou composts organiques. En conséquence, le taux de matière organique diminue. La matière organique constitue pourtant le substrat indispensable au développement de la vie biologique des sols, elle conditionne en fait leurs propriétés chimiques, physiques et biologiques. Le problème actuel grave de l'érosion des sols (érosion éolienne ou due au ruissellement des eaux) est pour une grande part une conséquence de teneurs insuffisantes en carbone organique. La matière organique constitue le substrat indispensable au développement de la vie biologique, car elle est la source majeure de carbone et d’énergie pour les micro-organismes. Comme nous le verrons, elle conditionne les propriétés chimiques (stocks de carbone, d’azote et de phosphore) et physiques (perméabilité, capacité de rétention et de circulation de l’eau, stabilité structurale). Il est donc indispensable de mieux connaître les processus de fonctionnement des sols pour mieux les utiliser et les préserver. En conséquence, il importe d’étudier la matière organique qu’ils contiennent. La matière organique des sols Origine de la matière organique des sols Au départ la photosynthèse des végétaux supérieurs ou inférieurs permet de transformer le carbone minéral, le gaz carbonique ou CO2, en carbone organique (Fig. 1). Les végétaux synthétisent ainsi leurs constituants, tissus divers, lignine, subérine etc. En milieu aquatique ou marin, le carbone minéral utilisable est sous forme carbonate (CO32-) ou hydrogénocarbonate (HCO3-) en équilibre avec CO2. Il faut noter que les végétaux sont incapables d’utiliser ou de transformer leurs déchets ou leurs débris organiques. Ce sont des autotrophes qui puisent dans le monde minéral ce qui leur faut pour réaliser la synthèse des molécules nécessaires à leur fonctionnement. Le nécessaire recyclage des matières premières implique l’existence d’hétérotrophes qui peuvent utiliser la matière organique déjà élaborée comme matière première et comme source d’énergie (Fig. 1). C’est la notion de chaîne alimentaire, les végétaux sont des producteurs, interviennent ensuite les consommateurs et les décomposeurs. La respiration des divers acteurs y compris les micro-organismes du sol réinjecte du gaz carbonique dans l’atmosphère. Dans le processus de décomposition des résidus animaux ou végétaux, une partie du carbone est assimilée dans les tissus microbiens, une autre part est transformée en humus stable. La décomposition des restes végétaux et animaux est une somme de processus biologiques qui permet par la minéralisation ou la respiration de réinjecter du carbone dans l’atmosphère sous forme de CO2. L’azote disponible est transformé en ions nitrate (NO3-) ou ammonium (NH4+), le soufre et le phosphore en ions sulfate (SO42-) et phosphate (PO43-) ou formes équivalentes assimilables par les plantes. Teneurs des sols en carbone organique La teneur en matière organique des sols varie fortement selon la nature des sols. Elle peut représenter de 5 à 6% du poids de sol sec pour une prairie (15 premiers centimètres) et moins de 1% pour les sols sableux. La teneur peut atteindre 10% pour des sols pauvres mal drainés (et 80 % ou plus dans les tourbières). Les sols tropicaux présentent des concentrations faibles, les conditions de température et d’humidité font que le turn-over de la matière organique est (trop) élevé. Les teneurs de carbone (C) et d’azote organique (N) varient généralement parallèlement (rapport C/N de 10 à 12 en moyenne). Il est intéressant de comparer les quantités de carbone présent dans les sols et dans d’autres réservoirs de la surface de la planète Terre. On peut constater qu’en surface la quantité de carbone organique (30 à 50 x 1014 kg) est supérieure à la somme des autres réservoirs (CO2 : 7 x 1014 kg; biomasse : 4,8 x 1014 kg; eaux douces : 2,5 x 1014 kg; C marin : 5 à 8 x 1014 kg. Un grand nombre de facteurs conditionnent la teneur en matière organique des sols. Parmi ces facteurs, on peut distinguer le climat qui conditionne la nature des plantes, la productivité végétale, l’activité microbienne. Les climats humides favorisent les forêts. Si l’humidité est suffisante la production végétale est accrue. Les sols de prairie renferment de grandes quantités d’humus dont la formation est importante dans la rhizosphère Si la température augmente, le taux d’humus décroît car il y a augmentation de l’activité microbienne. Ainsi, sous nos climats tempérés une litière sous forêt met une année pour se décomposer. Il n’y a pratiquement pas de litière sous climat tropical (conditions favorables à la biodégradation). Devenir du carbone organique dans les sols De nombreuses études ont tenté de préciser le devenir du carbone organique dans le sol en utilisant des substrats marqués au carbone 14. En se basant sur la seule cinétique, on peut distinguer trois catégories principales de matière organique : les résidus végétaux (ou la fumure apportée) et la biomasse associée qui se transforment en quelques années, les métabolites microbiens ou cellulaires et produits de transformation qui se "stabilisent" dans les sols et ont un turn-over de 5 à 25 ans et les fractions dites résistantes (devenue résistante ou originelle résistante) qui a un turn-over de 250 à 2500 ans. On arrive donc à la notion d’humus – ou de composés humiques – plus stables que les débris végétaux originels vis à vis de l’activité des micro-organismes du sol. Le processus est appelé humification. Cet humus n’est pas inerte dans les sols actifs, il se décompose peu à peu et correspond donc à un stock de carbone organique dans les sols. Agents responsables de la transformation du carbone organique dans les sols La décomposition et la transformation de la matière organique dans les sols sont assurées par un certain nombre d’organismes ou de micro-organismes. Tout d’abord les vers de terre et divers animaux du sol réduisent la taille des débris végétaux frais. Les vers de terre jouent un rôle très important, ils excrètent de la matière organique fine étroitement associée à la matière minérale (constituant ainsi une sorte de pré-humus; les termites produisent un matériau voisin des termites pour la construction des termitières). Il y a ensuite attaque microbienne (bactéries, champignons etc.). Les substances organiques les plus facilement décomposables (sucres…) sont tout d’abord utilisées. Le carbone est en partie métabolisé – il sert d’énergie – en partie utilisé pour la synthèse cellulaire (de 10 à 20% selon le sol et les conditions climatiques) – il sert donc de matière première - une autre partie est transformée peu à peu en humus. A côté des vers de terre et autres animaux du sol, en nombre très variable, les agents assurant la transformation de la matière organique sont les bactéries (1 milliard par gramme de sol, voire davantage), les actinomycètes[1] (plusieurs centaines de millions par gramme de sol), les champignons (de 10 à 20 millions par gramme de sol), les algues et les protozoaires. Âge moyen du carbone organique. Notion de temps de résidence Il est bien sûr tentant de déterminer un âge moyen de la matière organique dans un sol donné, c’est la notion de temps de résidence. Ceci est difficile, les âges absolus ne peuvent être déterminés, il y a en effet décomposition continuelle de l’humus ancien et synthèse parallèle d’humus nouveau. On ne peut déterminer qu’un âge moyen. Les diverses études conduites sur ce point montrent une très forte variabilité, de 250 à 1900 ans pour des horizons de surface. Le temps de résidence moyen augmente avec la profondeur. A titre d’exemple, pour un sol du Canada, le temps résidence moyen est de 8400 ans en profondeur, à comparer à 550 ans pour le niveau de surface. Le temps de résidence varie également selon la nature de la matière organique, l’humine étant généralement plus résistante que les lipides (ces catégories seront définies dans la suite de l’exposé). Ainsi dans un sol de prairie du Dakota, l’humine a un temps moyen de 1150 ans, alors que le temps moyen pour la matière organique totale est de 870 ans : ceci indique la très grande diversité, l’hétérogénéité de la matière organique. Les fraction de matière organique associées aux argiles ont généralement des temps de résidence élevés, par exemple 8000 ans. La matière organique adsorbée, ou incluse dans les feuillets de l’argile est protégée de l’activité biologique. Rôle de la matière organique dans les sols Propriétés chimiques La matière organique représente le stock de carbone, phosphore (P), azote (N) et soufre (S) présent dans le sol. Comme nous l’avons vu, ce stock est nécessaire pour toutes les formes de vie présentes sur (végétaux) et dans (microorganismes divers) le sol. Pour l’azote (N), le phosphore (P) et le soufre (S), il y a équilibre entre les formes minérales et organiques. Les processus qui interviennent sont la minéralisation et l’immobilisation (notion de stock). Minéralisation : P organique ¾® P minéral, soit phosphate (PO43-) S organique ¾® S minéral, soit sulfate (SO42-) N organique ¾® N minéral, soit nitrate (NO3-) Immobilisation : phosphate ¾® P organique sulfate ¾® S organique nitrate ¾® N organique Ces équilibres sont régis par de nombreux paramètres, de plus chaque élément majeur conditionne l’équilibre minéralisation – immobilisation de l’élément voisin (notion de rapport carbone/azote (C/N), carbone/phosphore (C/P), carbone/soufre (C/S) etc.]. Rappelons que seules les formes minérales peuvent être assimilées par les végétaux. Propriétés biologiques La matière organique est, comme nous l’avons vu source de carbone et d’énergie pour les micro-organismes lors de la synthèse de leur tissu par exemple. Il faut cependant noter que certaines molécules organiques peuvent jouer un rôle néfaste, par exemple certains terpènes naturels sous forêt de pin, ou des molécules issues de l’industrie chimique : rôle inhibiteur sur la germination des graines et sur la croissance des végétaux, toxicité envers la microflore; en conséquence la matière organique s’accumule trop, on arrive à la notion d’auto-intoxication dans les systèmes naturels pauvres. Propriétés physiques La matière organique est un facteur prépondérant pour la stabilité structurale du sol, elle joue le rôle de liant entre les particules, permettant la formation d’agrégats. Les principales interactions sont les associations minéraux-minéraux, minéraux-argile, argile-argile. Elle permet en outre une meilleure aération du sol (paramètre important pour l’activité biologique) et sa présence en quantité suffisante est un facteur important pour la capacité de rétention en eau du sol (importante pour les végétaux) et la circulation de l’eau dans le sol. On comprend alors mieux pourquoi les pratiques culturales modernes – agriculture intensive, monoculture, utilisation massive d’engrais minéraux azote, phosphore, potassium (N, P, K) au détriment de fumures ou composts organiques, désherbage systématique, déboisement et arasement des haies – ont des conséquences si néfastes sur les propriétés des sols. Les exportations de carbone dues aux récoltes ne sont plus compensées, en conséquence le taux de matière organique diminue. Ainsi, l’érosion des sols est devenue un problème préoccupant même en France. Il y a ainsi érosion éolienne et érosion mécanique sous l’effet des pluies; l’eau pénètre mal ou peu dans le sols et entraîne les particules de sol. Le problème est aggravé par la suppression de protections physiques (bois, haies, herbe qui de plus produisent du carbone organique à proximité de la zone de culture) et certaines pratiques comme le labour dans le sens de la pente. A titre d’exemple, le problème a été signalé récemment dans la presse nationale pour les vignes des coteaux du Sancerrois, aggravé par le désherbage systématique pratiqué aujourd’hui. Etude qualitative de la matière organique des sols Les formes persistantes (stables) de matière organique dans les sols sont les lipides et les substances humiques : acides fulviques, acides humiques et humine. Il est admis que les autres formes telles les protéines, les sucres… sont très rapidement décomposées et utilisées. Ces fractions sont obtenues en appliquant le protocole suivant (Fig. 2): - Les lipides dits libres correspondent à la fraction soluble dans un solvant organique, ils sont donc directement extraits par un solvant organique à partir de l’échantillon de sol. - Les lipides dits associés sont extraits après destruction des colloïdes organo-minéraux (argiles etc.) par un traitement acide chlorhydrique/acide fluorhydrique. - Les acides fulviques et humiques sont extraits en milieu basique (solution de soude). Ils ne sont donc pas solubles dans un solvant organique. En milieu acide, les acides humiques précipitent et sont donc séparés des acides fulviques qui restent solubles. - Le résidu final, associée à la matière minérale restante correspond à l’humine. Ainsi donc en résumé, les quatre grandes classes de MO sont les lipides solubles dans les solvants organiques usuels, les acides fulviques solubles en milieu aqueux à toute valeur de pH, les acides humiques solubles en milieu aqueux alcalin et l’humine totalement insoluble. Il est important de signaler que ces catégories ne correspondent pas à des classes précises de composés organiques, mais ne sont, dans l’état actuel des connaissances, définies que sur la base d’un protocole analytique (d’extraction). C’est une difficulté, une imprécision qui gène actuellement beaucoup les scientifiques. Les proportions de ces fractions organiques varient beaucoup d’un sol à un autre, les substances humiques sont particulièrement abondantes dans les sols tourbeux et les tourbes. Les substances humiques contiennent de 40 à 60% de carbone, 30 à 50% d’oxygène avec très peu d’azote de phosphore et de soufre. Les acides fulviques contiennent plus d’oxygène que les acides humiques. Acides humiques et humine ont des compositions proches, la totale insolubilité de l’humine est probablement due à une plus grande complexité structurale et à l’association avec la matière minérale, argile, métaux etc. Les lipides ont des structures plus simples et une gamme de poids moléculaire plus faible que les substances humiques. Pour celles-ci, le poids moléculaire varie de quelques centaines à plusieurs centaines de milliers d’unités de masse atomique. Quelques poids moléculaires sont donnés à titre de comparaison : eau : 18, gaz carbonique : 44, acide acétique (présent dans le vinaigre) : 60, cholestérol : 386, acide oléique [acide gras insaturé en C18 (possédant 18 atomes de carbone) présent dans les huiles alimentaires] : 282. Le poids moléculaire augmente dans le sens : acides fulviques → acides humiques → humine Les acides fulviques et humiques présents dans les eaux possèdent des poids moléculaires inférieurs à ceux présents dans le sol. Ainsi, ils peuvent circuler en phase aqueuse et sont donc des vecteurs importants, grâce à leurs propriétés complexantes, de propagation des pollutions, par exemple vers les nappes phréatiques à partir de la surface. Les lipides, plus hydrophobes, sont beaucoup moins mobiles ou mobilisables en phase aqueuse, mais ils migrent cependant, un certain nombre de classes contenant un groupe fonctionnel hydrophile (acide gras, alcool…). Mes lipides Les fractions de lipides "libres" ou "associés" contiennent, à un grand nombre de familles qui peuvent être séparées puis analysées principalement par des techniques de chromatographie en phase gazeuse capillaire, en phase liquide couplées à la spectrométrie de masse (ils sont appelés lipides simples). Ce n’est pas le cas des lipides complexes ou macromoléculaires qui ne peuvent être analysés directement (vide infra). Dans les familles qui contiennent un grand nombre de molécules, certains composés peuvent être spécifiques d’une origine donnée, on les appelle marqueurs. Les hydrocarbures (constitués uniquement de carbone et d’hydrogène) majeurs dans les sols possèdent un nombre impair d’atomes de carbone, majoritairement 29 et 31 atomes de carbone (C29, C31). Ils proviennent des tissus de protection des végétaux supérieurs. Pour les végétaux inférieurs tels les algues les hydrocarbures sont courts (C17). Les acides gras (acides monocarboxyliques) constituent une classe intéressante (Fig.3). Ils sont linéaires, majoritairement à nombre pair d’atomes de carbone, de C10 à C36. Ils sont saturés ou insaturés, une double liaison carbone-carbone remplaçant deux atomes d’hydrogène (acide oléique).
VIDEO CANAL U LIEN
( si la vidéo n'est pas visible,inscrivez le titre dans le moteur de recherche de CANAL U ) |
| |
|
| |
|
|
|
L'ION O2 |
|
|
| |
|
| |
LES CONFIGURATIONS ÉLECTRONIQUES DE L'ION O2- DANS DIVERS OXYDES MAGNÉTIQUES ET NON MAGNÉTIQUES : BEO, MGO, CAO, SRO, BAO, MNO, COO, NIO
Ce vidéo film est une étude de la distribution électronique dans divers oxydes dans lesquels l'oxygène est dans son état d'ionisation O2-. Pour chaque composé, on visualise 2 fois 2 types de représentations des densités différences, par rapport à un modèle uniquement composé d'atomes sphériques. A partir des études des cristaux, NiO, CoO et MnO, pour NiO, les ions Ni2+ et O2- forment leurs propres cages. Pour CoO, les déformations électroniques, autour de O2- , sont dues aux formations des liaisons entre les cages de O2- et le cation. Dans MnO, ces mêmes déformations sont moins prononcées. Pour les oxydes cubiques MgO, CaO, SrO et BaO, les déformations de O2- rendent l'ion complètement isolé du cation, amenant un caractère croissant de liaison ionique, depuis MgO jusqu'à BaO. L'étude de BeO montre que les distributions électroniques des ions O2- construisent des filets alternés-plans, de maille hexagonale. Des ponts, entre les différents niveaux des plans, enchâssent les ions Be2+ dans le réseau des O2-. Les cations jouent simplement le rôle d'agents électrostatiques stabilisants.
VIDEO CANAL U LIEN
(si la video n'est pas accéssible,tapez le titre dans le moteur de recherche de CANAL U.) |
| |
|
| |
|
| Page : [ 1 2 3 4 5 6 7 8 9 10 ] Précédente - Suivante |
|
|
|
|
|
|