|
|
|
|
 |
|
Les lymphocytes B : de nouveaux alliés pour le traitement des sarcomes par immunothérapie ? |
|
|
| |
|
| |

Les lymphocytes B : de nouveaux alliés pour le traitement des sarcomes par immunothérapie ?
COMMUNIQUÉ | 15 JANV. 2020 - 19H00 | PAR INSERM (SALLE DE PRESSE)
CANCER
Comment améliorer et mieux personnaliser les traitements des sarcomes des tissus mous, ces cancers agressifs et particulièrement résistants ? Une équipe internationale dirigée par Wolf Hervé Fridman et regroupant des chercheurs de l’Inserm, de Sorbonne Université et d’Université de Paris au Centre de recherche des Cordeliers, en collaboration avec la Ligue nationale contre le cancer et l’Institut Bergonié, montre que les lymphocytes B permettent de prédire la réponse des patients à l’immunothérapie, rôle que l’on croyait jusqu’à présent dévolu seulement aux lymphocytes T. Ces résultats à paraître dans Nature ouvrent la voie à la personnalisation des traitements pour les patients atteints de sarcomes des tissus mous.
Les sarcomes des tissus mous sont un groupe hétérogène de cancers agressifs et résistants à la chimiothérapie qui touchent les tissus mous de l’organisme (graisse, muscles, tissus fibreux, vaisseaux sanguins et lymphatiques, nerfs…). Dans les essais cliniques actuels, seuls 15 % des patients répondent à l’immunothérapie, ce qui pose la question de l’exposition inutile des autres patients à la toxicité de ces traitements. Identifier des marqueurs prédisant la réponse à l’immunothérapie est donc un enjeu primordial. Cependant, jusqu’à aujourd’hui, cette stratégie se focalisait essentiellement sur les lymphocytes T, les cellules immunitaires capables de reconnaître les cellules infectées, cancéreuses ou étrangères à l’organisme.
À travers des travaux publiés dans la revue Nature, une équipe de recherche menée par Wolf Hervé Fridman et regroupant des chercheurs de l’Inserm, de Sorbonne Université et d’Université de Paris au Centre de recherche des Cordeliers, en collaboration avec l’équipe Carte d’identité des tumeurs de la Ligue nationale contre le cancer, l’Institut Bergonié, et des équipes américaines et taïwanaises, s’est penchée sur la question de l’identification d’autres marqueurs potentiels.
Les chercheurs ont analysé 608 tumeurs, qu’ils ont classées en trois groupes suivant la composition de leur microenvironnement tumoral[1] : les tumeurs immunologiquement pauvres (pauvres en cellules immunitaires et peu vascularisées), les tumeurs fortement vascularisées et enfin les tumeurs immunologiquement riches. Ces dernières présentent des agrégats de différents types cellulaires riches en lymphocytes B, les cellules immunitaires responsables de la production d’anticorps. Ces agrégats sont appelés structures lymphoïdes tertiaires. Les chercheurs ont observé qu’une réponse immunitaire antitumorale s’initiait en leur sein, montrant par-là que les lymphocytes B pourraient jouer un rôle antitumoral.
De plus, dans un essai clinique de phase 2, les patients présentant des tumeurs immunologiquement riches ont montré un taux de réponse élevé (50 %) à une immunothérapie : le pembrolizumab. Ces patients avaient en outre un taux de survie plus élevé que ceux présentant des tumeurs immunologiquement pauvres ou fortement vascularisées.
Une seconde étude d’une équipe américaine, cosignée par l’équipe de Wolf Hervé Fridman au Centre de recherche des Cordeliers (Inserm/Sorbonne Université/Université de Paris) et publiée en parallèle dans Nature, a permis d’étendre ces observations au mélanome et au cancer du rein.
Les résultats de ces études montrent qu’en plus des lymphocytes T habituellement étudiés, les lymphocytes B seraient essentiels dans la réponse à l’immunothérapie pour certains cancers. Ils apportent un nouvel espoir pour le traitement des sarcomes des tissus mous, cancers particulièrement résistants aux thérapies classiques. De plus, dans un objectif de médecine personnalisée, ces résultats pourraient aider à guider la prise de décision clinique et le traitement des patients grâce à un simple test permettant d’identifier ceux ayant des tumeurs immunologiquement riches.
Sur la base de ces résultats, un premier essai clinique français coordonné par Antoine Italiano (Institut Bergonié, Université de Bordeaux), co-auteur du premier article, et incluant des patients présentant des tumeurs immunologiquement riches est actuellement en cours au sein du Groupe Sarcome Français.
[1] Le microenvironnement tumoral correspond aux éléments biologiques entourant la tumeur (vaisseaux sanguins, cellules immunitaires, types variés de cellules, molécules de signalisation, matrice extracellulaire…) et avec lesquels elle interagit.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Une nouvelle canalopathie cérébrale associant déficience intellectuelle et mouvements anormaux |
|
|
| |
|
| |
Une nouvelle canalopathie cérébrale associant déficience intellectuelle et mouvements anormaux
COMMUNIQUÉ | 27 NOV. 2020 - 9H00 | PAR INSERM (SALLE DE PRESSE)
NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Les dysfonctionnements des canaux ioniques – ou canalopathies – dans le cerveau sont aujourd’hui associés à plus de 30 maladies neurologiques comme l’épilepsie ou encore les ataxies cérébelleuses. Structures situées sur la membrane des cellules permettant le passage d’ions (par exemple les ions sodium et potassium) entre l’intérieur d’une cellule et son environnement extérieur (milieu extracellulaire), ces canaux permettent notamment de générer et contrôler les potentiels d’action dans les neurones. Une étude menée à l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) a permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Les résultats ont été publiés dans Brain le 27 novembre 2020.
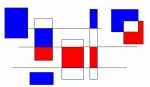
Les variants pathogéniques du gène KCNN2 identifiés chez les patients et leur localisation sur la structure protéique du canal SK2.
Les variant en rouge sont des variants pathogènes tronquant (introduisant un codon stop dans la séquence protéique). Les variants en noirs sont les variants pathogènes faux-sens associés à une perte de fonction. Le variant en gris a été classé de signification inconnue car le canal avec ce variant n’a pas montré de déficit particulier en électrophysiologie.
Le Dr Fanny Mochel, généticienne au sein du département de génétique de l’hôpital de la Pitié-Salpêtrière AP-HP et chercheuse à l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) et le Pr Christel Depienne, généticienne à l’institut de génétique humaine de l’Hôpital Universitaire d’Essen (Allemagne) et également chercheuse à l’Institut du cerveau ont identifié un nouveau syndrome associé à des mutations du canal SK2. L’étude publiée dans la revue scientifique Brain porte sur 10 patients, 6 hommes et 4 femmes âgés de 2 à 60 ans présentant des retards intellectuels plus ou moins sévères associés, pour certains, à des troubles du spectre autistique ou des épisodes psychotiques. Ces troubles cognitifs sont dans tous les cas associés à des tremblements, à des symptômes d’ataxie cérébelleuse ou encore à des mouvements anormaux.
Grâce à une collaboration avec Agnès Rastetter de la plateforme de génotypage/séquençage de l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS), le génome d’un premier patient recruté à la Pitié-Salpêtrière a été analysé à la recherche de mutations génétiques à l’origine de ce syndrome. Cette analyse a mis en évidence une mutation du gène KCNN2 interrompant sa séquence codante, absente des parents du patient (mutation de novo). L’imagerie cérébrale par IRM (imagerie par résonance magnétique) chez ce patient a mis en évidence des anomalies de structure et d’intégrité de la substance blanche du cerveau, c’est-à-dire la gaine cérébrale protectrice des axones des neurones.
Par ailleurs, une collaboration internationale a permis aux chercheurs d’identifier 9 autres patients avec mutations du gène KCNN2. La majorité de ces mutations étaient survenues de novo tandis qu’une mutation était transmise dans une forme familiale du même syndrome.
Enfin, en travaillant conjointement avec Carine Dalle de la plateforme d’exploration cellulaire d’électrophysiologie de l’Institut du cerveau, les équipes des Dr Mochel et Depienne ont montré un rôle délétère de ces mutations sur la fonction du canal SK2, c’est-à-dire une perte de fonction entrainant un dysfonctionnement du canal ionique SK2 et donc une perte de régulation du potentiel d’action, support du message nerveux.
Les résultats de cette nouvelle étude ont permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Ce nouveau syndrome se caractérise par la présence, d’une part, de symptômes cognitifs, en particulier une déficience intellectuelle et, d’autre part, de symptômes moteurs tels que des mouvements anormaux.
Cette nouvelle pathologie, dont on connaît maintenant la cause, est très hétérogène d’un point de vue des symptômes et nécessite une prise en charge multidisciplinaire à la frontière entre la génétique, pour la recherche des mutations du gène KCNN2, la neuropédiatrie et la neurologie pour la prise en charge des manifestations cognitives et motrices des patients.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Les agrégats de méningocoques coulent dans les vaisseaux sanguins comme un liquide visqueux |
|
|
| |
|
| |
Les agrégats de méningocoques coulent dans les vaisseaux sanguins comme un liquide visqueux
vendredi 6 juillet 2018
Une collaboration entre biologistes et physiciens a permis de décrypter une étape clé de l’infection causée par le méningocoque, un pathogène humain responsable de méningites chez les nourrissons et les jeunes adultes, pathologies qui, malgré une prise en charge rapide, présentent un taux de mortalité qui reste très important. Ce travail a été publié le 17 mai 2018, dans la revue Cell.
L’infection humaine se caractérise par l’accumulation de bactéries à l’intérieur des vaisseaux sanguins qui se trouvent entièrement remplis de bactéries bien que, ni les mécanismes de formation, ni l’impact de ce processus ne soient connus. Intrigués par la formation de ces agrégats intravasculaire, le consortium de scientifiques, regroupant biologistes et physiciens, s’est attelé à comprendre cette étape de l’infection, tout particulièrement sa base physique.
Il ressort de cette étude que les agrégats bactériens formés par le méningocoque se comportent de façon inattendue, comme un liquide visqueux, avec la viscosité du miel. Les bactéries se multiplient rapidement dans les vaisseaux sous forme d’agrégats qui s’adaptent ainsi progressivement à la géométrie complexe du réseau vasculaire, comme un liquide qui s’écoule. L’étude montre que la formation de ces agrégats et leurs propriétés physiques sont essentielles pour la progression de l’infection.
Les propriétés de liquide visqueux des agrégats dépendent d’un facteur de virulence appelé pilus de type IV. Il s’agit de long filaments adhésifs et dynamiques qui s’allongent et se rétractent en permanence à la surface de la bactérie. Ces filaments permettent aux bactéries de se trouver, de se rapprocher et d’entrer en contact de façon réversible. L’agrégation est donc basée sur un processus aléatoire d’attraction entre les bactéries. Ces filaments permettent aux bactéries de se trouver, de se rapprocher et d’entrer en contact de façon réversible. L’observation montre que cette dynamique d’interaction entre bactéries voisines est aléatoire, et contribue donc de manière active à l’agitation globale des bactéries dans l’agrégat. Sur le plan physique ce processus d’interaction confère à ces agrégats des propriétés originales de fluide hors d’équilibre jusque-là non décrites. Par exemple, les bactéries à l’intérieur des agrégats présentent une motilité plus élevée que celle observée par la diffusion des bactéries isolées. Ainsi, au-delà de proposer une meilleure compréhension d’une infection humaine létale, cette étude dévoile un nouveau type de matière active basée sur la présence des forces attractives intermittentes entre ses éléments constituants.
Cette étude pluridisciplinaire a pu être réalisée grâce à une étroite collaboration entre un laboratoire spécialisé dans les infections causées par le méningocoque (G. Duménil, Institut Pasteur et INSERM) et des physiciens. La collaboration avec les équipes de Hugues Chaté (CEA, CNRS, Université Paris-Saclay), Nelly Henry (CNRS, Sorbonne Université) et Raphael Voituriez (CNRS, Sorbonne Université) a permis de coupler une approche expérimentale quantitative avec un modèle physique de matière active.
Figure : Vue d’artiste d’un agrégat de bactéries Neisseria meningitidis aspiré par une micropipette. Sous application d’une pression négative, ce matériau présente des propriétés uniques de fluide visqueux, dues à des forces d’interactions intermittentes entre bactéries engendrées par des pili de type IV (représentés par des filaments colorés). La fluidité des agrégats est essentielle pour une colonisation efficace du réseau vasculaire.
© Julien Husson (https://cellmechanics.jimdo.com).
Références :
* Intermittent Pili-Mediated Forces Fluidize Neisseria meningitidis Aggregates Promoting Vascular Colonization.
Bonazzi D, Lo Schiavo V, Machata S, Djafer-Cherif I, Nivoit P, Manriquez V, Tanimoto H, Husson J, Henry N, Chaté H, Voituriez R, Duménil G.
Cell. 2018 Jun 28;174(1):143-155.e16. doi: 10.1016/j.cell.2018.04.010. Epub 2018 May 17.
*
Contacts :
* Raphael Voituriez
Tél. 33 (0)1 44 27 37 03
Laboratoire Jean Perrin
CNRS/Sorbonne université
4 place Jussieu, 75005 PARIS
DOCUMENT CNRS LIEN
|
| |
|
| |
|
|
|
L'ADN et la médecine génomique personnalisée |
|
|
| |
|
| |
L'ADN et la médecine génomique personnalisée
Publié le 30 novembre 2017
*
Chaque individu est unique. Les particularités de chacun sont essentiellement explicables par l’ADN qui, comme un plan d’architecte, codifie tout notre organisme. Mais il n’est pas figé et peut évoluer sous l’influence de son environnement. L’ADN code également les risques de développer certaines maladies. Il impacte l’efficacité de certains traitements, notamment contre le cancer. Sa prise en compte dans la médecine constitue un enjeu majeur pour adapter les traitements en fonction des dispositions génétiques de chacun. C’est ce qu’on appelle la médecine génomique personnalisée. Mais la connaissance de l’ADN des individus n’est pas le seul facteur à considérer pour comprendre notre organisme. De plus, elle pose de nombreuses questions éthiques.
QU’EST-CE QUE L’ADN ?
L’ADN se trouve dans la plus petite unité du vivant : la cellule. En son cœur est rassemblé l’ensemble de nos caractères héréditaires : le génome. Une molécule d’ADN ressemble à une échelle qui s’enroule sur elle-même. Chaque barreau de cette échelle est constitué de deux petites molécules différentes, appelées bases ou nucléotides. On en dénombre 4 différentes : adénine (A), thymine (T), cytosine (C) et guanine (G). Elles sont dites complémentaires car elles s’apparient toujours de la même façon (A avec T et C avec G). Ce code génétique est universel à tous les êtres vivants.
Déroulé, l’ADN mesurerait 1 mètre de haut et serait 1 000 fois plus fin qu’un cheveu. Lors de la division cellulaire, l’ADN se compacte et s’organise en bâtonnets, appelés chromosomes. Leur nombre varie d’une espèce à l’autre. L'Homme possède 46 chromosomes répartis en 23 paires : 22 paires d'autosomes et 1 paire de gonosomes ou chromosomes sexuels, appelés X et Y. Les hommes possèdent un chromosome X et un chromosome Y. Les femmes possèdent 2 chromosomes X. Par comparaison, le riz possède 24 chromosomes et la mouche 8. L’analyse des chromosomes humains permet par exemple de connaître le sexe d’un individu ou de déceler certaines maladies génétiques comme la trisomie 21 (possession de 3 copies du chromosome 21).
Historique des découvertes et avancées sur l’ADN et la génétique
La découverte de la structure de l’ADN a lieu dans les années 50. Il faudra pourtant attendre 2003 pour réussir à déchiffrer entièrement tout l’ADN du génome humain. Aujourd’hui les progrès réalisés dans les technologies de séquençage permettent de développer une médecine génomique personnalisée.
Découvrez en animation-vidéo l’histoire des principales avancées dans le domaine de l’ADN et de la génétique.
Vidéo
La découverte de l'ADN
<div class="reponse warning"> <p>Pour accéder à toutes les fonctionnalités de ce site, vous devez activer JavaScript. Voici les <a href="http://www.enable-javascript.com/fr/">instructions pour activer JavaScript dans votre navigateur Web</a>.</p> </div> VOIR DANS LA MÉDIATHÈQUE
A quoi sert l’ADN ?
Certains enchainements de nucléotides dans l’ADN fournissent des instructions qui commandent la synthèse de protéines ; ce sont les gènes. Unités de base de l’hérédité, ils déterminent ce que nous sommes et comment nous fonctionnons (couleur des yeux, groupe sanguin…). Il en existe environ 21 000 chez l’Homme. La plupart des gènes code une protéine et le rôle qu’elle jouera dans l’organisme. Certaines participent au transport, à la signalisation cellulaire… D’autres, comme les enzymes, catalysent des réactions chimiques. Les gènes peuvent être comparés aux parties d’un plan d’un architecte de notre organisme.
QU’EST-CE QUE L’ÉPIGÉNÉTIQUE ?
Comment expliquer la différence entre une cellule du foie et un neurone alors que toutes les deux renferment le même patrimoine génétique ? Par l’épigénétique, science qui établit le lien entre les caractères observables (phénotypes) et l’ensemble des gènes (génotypes). Pour faire un parallèle, si l’organisme vivant était une voiture ; la génétique serait l’établi sur lequel sont exposées toutes les pièces mécaniques et l’épigénétique la chaîne d’assemblage des différents éléments. Ainsi, l’épigénétique joue le rôle de chef d’orchestre en indiquant pour chaque gène à quel moment et dans quel tissu il doit s’exprimer.
Suite à la découverte des premiers mécanismes épigénétiques qui régulent l’expression des gènes, les chercheurs ont appris à désactiver l’expression d’un gène à des fins thérapeutiques.
Complémentaire à la génétique, l’épigénétique donne une vue plus complète de la machinerie cellulaire et révèle une surprenante complexité dans les régulations de l’expression génique. Elle ouvre des perspectives dans la compréhension et le traitement de nombreuses maladies.
QU’EST-CE QU’UNE MUTATION DE L’ADN ?
L’ADN code tous les organismes vivants. Avec le temps, il peut évoluer lors de la création de nouvelles cellules ou en réponse à son environnement.
Lorsqu’elle se divise, la cellule déclenche le processus de réplication de l’ADN pour en obtenir une copie. De temps en temps, le système produit quelques erreurs : ce sont les mutations. Le plus souvent, elles sont sans conséquence, puisqu’elles ont lieu dans les 98% du génome qui ne codent pas pour la synthèse d’une protéine (ADN non-codant ayant d’autres fonctions comme la régulation de l’expression des gènes).
D’autres mutations, en revanche, peuvent modifier la composition ou la quantité d’une protéine et être à l’origine d’une maladie génétique.
D’autres sources, environnementales ou liées aux activités de l’Homme, peuvent également modifier l’ADN.
Les facteurs mutagènes sont :
* Biologiques. Dans la nature il existe des agents biologiques particulièrement efficaces, les virus, dont certains peuvent tuer.
*
* Physiques. Les rayons UV, X et la radioactivité sont des agents physiques qui adoptent une méthode radicale : ils cassent la molécule d’ADN.
*
* Chimiques. Ils sont très nombreux, par exemple : le benzopyrène, présent dans la fumée de cigarette, le trichloréthylène, utilisé comme solvant dans les pressings...
QU’EST-CE QUE LA MÉTAGÉNOMIQUE ?
Le génome donne de nombreuses informations sur un individu. Cependant, le fonctionnement de nos cellules et de notre corps est également influencé par les quelques centaines de milliards de bactéries qui le colonisent. L’ADN de toutes ces bactéries correspond au métagénome.
L’analyse du métagénome d’un individu est importante car celui-ci influence le développement de certaines maladies comme le diabète, l’obésité ou encore certains cancers. De nouvelles thérapies reposent sur la métagénomique pour soigner certains cancers.
LA MÉDECINE GÉNOMIQUE PERSONNALISÉE
Identifier les gènes responsables de certaines maladies
Pour mieux soigner les maladies, il est nécessaire de connaître leurs causes. Analyser l’ADN pour trouver les gènes qui en sont responsables permet des diagnostics et pronostics (prévision de l’évolution d’une maladie) plus sûrs.
En 2016, les gènes responsables de plus de la moitié des 7 275 maladies monogéniques (maladie provoquée par la mutation d’un seul gène) recensées ont été identifiés. Ces performances ont été rendues possibles grâce au perfectionnement en temps et coût du séquençage et du génotypage de l’ADN. Pour que cette identification soit pertinente, il est nécessaire de rassembler une importante base de données de génomes de personnes saines ou malades afin de séquencer, analyser et comparer les données.
Adapter les traitements aux gènes des individus
Depuis peu, la médecine se rend compte des limites de donner le même traitement à différents patients atteints d’une même maladie. Les taux de réponse aux traitements traditionnels varient entre 20 et 80 %. Les différences génétiques individuelles peuvent être plus ou moins responsables de l’efficacité des traitements.
Dans le cadre du traitement du cancer, les différents traitements possibles pourront être testés sur les cellules tumorales du patient. Séquencer les tumeurs peut également permettre de trouver le traitement le plus efficace.
En juin 2016, la France se lance officiellement dans la bataille mondiale de la médecine génomique personnalisée avec le plan « France Médecine Génomique 2025 ». Ce dernier vise notamment à intégrer le séquençage de l’ADN dans la prise en charge des patients. Pour cela, le plan prévoit de déployer un réseau de douze plateformes de séquençage à haut débit du génome couvrant l’ensemble du territoire.
LES ENJEUX DE LA MÉDECINE GÉNOMIQUE PERSONNALISÉE
La médecine génomique suscite de nombreux espoirs. A court/moyen terme, elle révolutionnera la médecine en donnant les bons traitements ciblés directement sur le patrimoine génétique. A plus long terme, elle permettra également, en comprenant les mécanismes génétiques à l’origine des maladies monogéniques, de développer de nouvelles thérapies qui corrigent l’ADN pour soigner les cellules malades.
Le développement de la médecine génomique personnalisée pose également de nombreuses questions pratiques et éthiques.
D’un point de vue pratique :
* Pour fonctionner correctement, la médecine génomique nécessite un nombre important de séquençages d’ADN de personnes saines et malades. C’est uniquement par de larges études qu’il sera possible d’identifier les marqueurs génétiques qui permettront de proposer des traitements adaptés aux patients.
*
* La seule compréhension du génome humain ne suffit pas. L’être humain est un écosystème constitué de son génome mais aussi du génome des bactéries qui le colonisent. Pour progresser dans la médecine génomique personnalisée, il faut également prendre en compte la métagénomique et l’épigénétique (mécanismes qui agissent sur l’expression de l’ADN). Là encore, l’analyse poussée des données d’un grand nombre de patients sera nécessaire.
*
* L’entrée de la génétique dans l’ère du big data requiert l’acquisition de supercalculateurs et d’algorithmes pour pouvoir traiter d’énormes bases de données.
*
* La sécurité des données est un dernier enjeu si on cherche à constituer une importante base de données de génomes humains.
*
*
D’un point de vue éthique :
* Les pratiques doivent être encadrées afin d’éviter certaines dérives comme le choix de gènes ou de gamètes lors de la procréation médicalement assistée par exemple.
*
* L’analyse de l’ADN permet de connaître les prédispositions génétiques d’un individu sur de nombreuses maladies. Mais on peut se demander s’il est préférable de vivre dans l’ignorance ou de connaître les risques de développer une maladie génétique ? Actuellement, en France, seuls des tests ciblés sur des gènes qui pourraient être responsables de maladies sont réalisés sur prescription médicale.
DOCUMENT cea LIEN
|
| |
|
| |
|
| Page : [ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 ] Précédente - Suivante |
|
|
|
|
|
|